Portfolio
My classwork from bimm143
Class11: AlphaFold
Samuel Fisher (A18131929)
Background
In this hands-on session we will utilize AlphaFold to predict protein structure from sequence (Jumper et al. 2021). Without the aid of such approaches, it can take years of expensive laboratory work to determine the structure of just one protein. With AlphaFold we can now accurately compute a typical protein structure in as little as ten minutes. The PDB database(the main repositry of experimental structures) only has approximately 250 thousand structures (we saw this in the last lab). The main protein sequence database has over 200 million sequences! Only 0.125% of known sequences have a known structure - this is called the “structure knowledge gap”.
(250000 / 200000000) * 100
[1] 0.125
Structures are much harder to determine than sequences They are expensive (on average ~$1 million each) They take on average 3-5 years to solve!
EBI AlphaFold Database
The EBI has a database of pre-computed AlphaFold (AF) models called AFDB. This is growing all the time and can be useful to check before running AF ourselves.
Running AlphaFold
We can download and run locally (on our own computers) but we need a GPU. Or we can use “cloud” computing to run this on somene else’s computer
We will use ColabFold < https://github.com/sokrypton/ColabFold
We previously found there was no AFDB entry for our HIV sequence.
>HIV-Pr-Dimer
PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF:PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIGGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
Here we will use AlphaFold2_mmseqs2
I was unable to use Mol* to generate the image frm section 7. I was able to view the proteins, but unable to select the individual proteins in order to superimpose them.
Section 8
Section 8: point to YOUR results folder (must contain the 5 .pdb files)
results_dir <- "hivpr_23119/"
## list all PDB models in current project folder
pdb_files <- list.files(pattern="\\.pdb$", full.names=TRUE)
basename(pdb_files)
[1] "hivpr_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb"
[2] "hivpr_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb"
[3] "hivpr_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb"
[4] "hivpr_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb"
[5] "hivpr_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb"
[6] "m1_conserv.pdb"
library(bio3d)
# Read all PDB models and superpose/fit coordinates
pdbs <- pdbaln(pdb_files, fit=TRUE, exefile="msa")
Reading PDB files:
./hivpr_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb
./hivpr_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb
./hivpr_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb
./hivpr_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb
./hivpr_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb
./m1_conserv.pdb
......
Extracting sequences
pdb/seq: 1 name: ./hivpr_23119_unrelaxed_rank_001_alphafold2_multimer_v3_model_4_seed_000.pdb
pdb/seq: 2 name: ./hivpr_23119_unrelaxed_rank_002_alphafold2_multimer_v3_model_1_seed_000.pdb
pdb/seq: 3 name: ./hivpr_23119_unrelaxed_rank_003_alphafold2_multimer_v3_model_5_seed_000.pdb
pdb/seq: 4 name: ./hivpr_23119_unrelaxed_rank_004_alphafold2_multimer_v3_model_2_seed_000.pdb
pdb/seq: 5 name: ./hivpr_23119_unrelaxed_rank_005_alphafold2_multimer_v3_model_3_seed_000.pdb
pdb/seq: 6 name: ./m1_conserv.pdb
pdbs
1 . . . . 50
[Truncated_Name:1]hivpr_2311 PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:2]hivpr_2311 PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:3]hivpr_2311 PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:4]hivpr_2311 PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:5]hivpr_2311 PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
[Truncated_Name:6]m1_conserv PQITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGI
**************************************************
1 . . . . 50
51 . . . . 100
[Truncated_Name:1]hivpr_2311 GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:2]hivpr_2311 GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:3]hivpr_2311 GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:4]hivpr_2311 GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:5]hivpr_2311 GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
[Truncated_Name:6]m1_conserv GGFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNFP
**************************************************
51 . . . . 100
101 . . . . 150
[Truncated_Name:1]hivpr_2311 QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:2]hivpr_2311 QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:3]hivpr_2311 QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:4]hivpr_2311 QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:5]hivpr_2311 QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
[Truncated_Name:6]m1_conserv QITLWQRPLVTIKIGGQLKEALLDTGADDTVLEEMSLPGRWKPKMIGGIG
**************************************************
101 . . . . 150
151 . . . . 198
[Truncated_Name:1]hivpr_2311 GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:2]hivpr_2311 GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:3]hivpr_2311 GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:4]hivpr_2311 GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:5]hivpr_2311 GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
[Truncated_Name:6]m1_conserv GFIKVRQYDQILIEICGHKAIGTVLVGPTPVNIIGRNLLTQIGCTLNF
************************************************
151 . . . . 198
Call:
pdbaln(files = pdb_files, fit = TRUE, exefile = "msa")
Class:
pdbs, fasta
Alignment dimensions:
6 sequence rows; 198 position columns (198 non-gap, 0 gap)
+ attr: xyz, resno, b, chain, id, ali, resid, sse, call
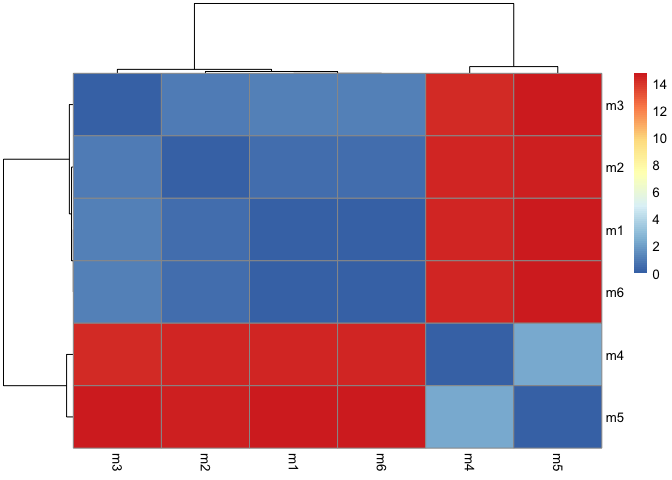
rd <- rmsd(pdbs, fit=TRUE)
Warning in rmsd(pdbs, fit = TRUE): No indices provided, using the 198 non NA positions
range(rd)
[1] 0.000 14.754
library(pheatmap)
# compute RMSD matrix
rd <- rmsd(pdbs, fit = TRUE)
Warning in rmsd(pdbs, fit = TRUE): No indices provided, using the 198 non NA positions
# auto-size labels to match matrix
n <- ncol(rd)
labs <- paste0("m", seq_len(n))
colnames(rd) <- labs
rownames(rd) <- labs
pheatmap(rd)
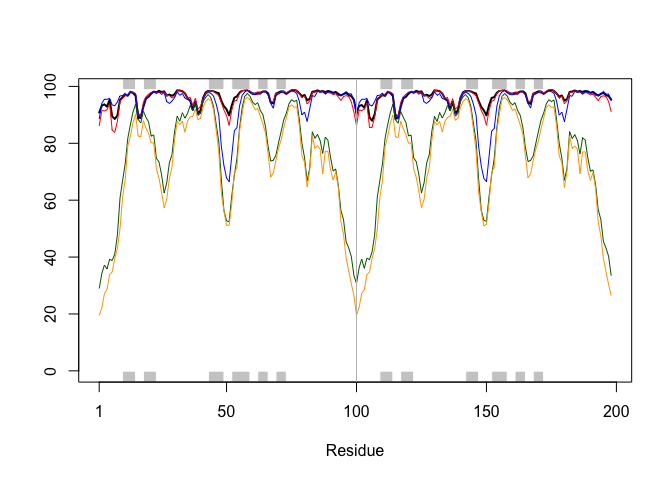
library(bio3d)
# Read a reference PDB for secondary structure annotation
pdb <- read.pdb("1hsg")
Note: Accessing on-line PDB file
plotb3(pdbs$b[1,], typ="l", lwd=2, sse=pdb)
points(pdbs$b[2,], typ="l", col="red")
points(pdbs$b[3,], typ="l", col="blue")
points(pdbs$b[4,], typ="l", col="darkgreen")
points(pdbs$b[5,], typ="l", col="orange")
abline(v=100, col="gray")
core <- core.find(pdbs)
core size 197 of 198 vol = 8198.676
core size 196 of 198 vol = 6466.264
core size 195 of 198 vol = 1439.037
core size 194 of 198 vol = 988.633
core size 193 of 198 vol = 887.094
core size 192 of 198 vol = 838.613
core size 191 of 198 vol = 792.884
core size 190 of 198 vol = 752.45
core size 189 of 198 vol = 712.61
core size 188 of 198 vol = 672.479
core size 187 of 198 vol = 637.04
core size 186 of 198 vol = 603.513
core size 185 of 198 vol = 561.938
core size 184 of 198 vol = 521.492
core size 183 of 198 vol = 490.743
core size 182 of 198 vol = 470.17
core size 181 of 198 vol = 450.848
core size 180 of 198 vol = 431.1
core size 179 of 198 vol = 411.439
core size 178 of 198 vol = 393.88
core size 177 of 198 vol = 379.206
core size 176 of 198 vol = 365.7
core size 175 of 198 vol = 352.437
core size 174 of 198 vol = 341.782
core size 173 of 198 vol = 334.26
core size 172 of 198 vol = 323.212
core size 171 of 198 vol = 313.806
core size 170 of 198 vol = 302.238
core size 169 of 198 vol = 290.89
core size 168 of 198 vol = 280.624
core size 167 of 198 vol = 266.73
core size 166 of 198 vol = 257.645
core size 165 of 198 vol = 249.157
core size 164 of 198 vol = 243.099
core size 163 of 198 vol = 235.451
core size 162 of 198 vol = 224.741
core size 161 of 198 vol = 218.717
core size 160 of 198 vol = 212.151
core size 159 of 198 vol = 205.394
core size 158 of 198 vol = 198.075
core size 157 of 198 vol = 194.178
core size 156 of 198 vol = 185.737
core size 155 of 198 vol = 180.836
core size 154 of 198 vol = 174.762
core size 153 of 198 vol = 170.987
core size 152 of 198 vol = 164.757
core size 151 of 198 vol = 158.084
core size 150 of 198 vol = 152.363
core size 149 of 198 vol = 146.502
core size 148 of 198 vol = 141.615
core size 147 of 198 vol = 134.788
core size 146 of 198 vol = 129.426
core size 145 of 198 vol = 123.661
core size 144 of 198 vol = 119.002
core size 143 of 198 vol = 113.911
core size 142 of 198 vol = 109.199
core size 141 of 198 vol = 103.485
core size 140 of 198 vol = 98.718
core size 139 of 198 vol = 94.56
core size 138 of 198 vol = 91.151
core size 137 of 198 vol = 88.428
core size 136 of 198 vol = 85.16
core size 135 of 198 vol = 82.251
core size 134 of 198 vol = 80.234
core size 133 of 198 vol = 77.194
core size 132 of 198 vol = 73.33
core size 131 of 198 vol = 70.103
core size 130 of 198 vol = 68.973
core size 129 of 198 vol = 67.193
core size 128 of 198 vol = 65.204
core size 127 of 198 vol = 62.351
core size 126 of 198 vol = 60.125
core size 125 of 198 vol = 58.102
core size 124 of 198 vol = 55.048
core size 123 of 198 vol = 53.226
core size 122 of 198 vol = 51.658
core size 121 of 198 vol = 49.896
core size 120 of 198 vol = 47.425
core size 119 of 198 vol = 45.513
core size 118 of 198 vol = 43.55
core size 117 of 198 vol = 41.596
core size 116 of 198 vol = 39.959
core size 115 of 198 vol = 37.995
core size 114 of 198 vol = 36.305
core size 113 of 198 vol = 34.012
core size 112 of 198 vol = 32.587
core size 111 of 198 vol = 30.936
core size 110 of 198 vol = 28.592
core size 109 of 198 vol = 26.311
core size 108 of 198 vol = 24.595
core size 107 of 198 vol = 22.869
core size 106 of 198 vol = 21.533
core size 105 of 198 vol = 20.217
core size 104 of 198 vol = 18.957
core size 103 of 198 vol = 17.506
core size 102 of 198 vol = 15.686
core size 101 of 198 vol = 14.273
core size 100 of 198 vol = 13.514
core size 99 of 198 vol = 10.915
core size 98 of 198 vol = 9.077
core size 97 of 198 vol = 7.544
core size 96 of 198 vol = 5.89
core size 95 of 198 vol = 5.263
core size 94 of 198 vol = 4.805
core size 93 of 198 vol = 3.981
core size 92 of 198 vol = 3.133
core size 91 of 198 vol = 2.4
core size 90 of 198 vol = 1.861
core size 89 of 198 vol = 1.485
core size 88 of 198 vol = 0.966
core size 87 of 198 vol = 0.717
core size 86 of 198 vol = 0.574
core size 85 of 198 vol = 0.445
FINISHED: Min vol ( 0.5 ) reached
core.inds <- print(core, vol=0.5)
# 86 positions (cumulative volume <= 0.5 Angstrom^3)
start end length
1 9 50 42
2 52 95 44
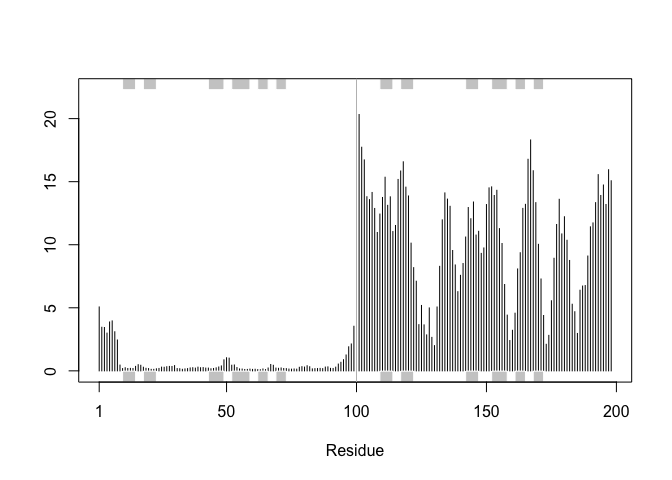
xyz <- pdbfit(pdbs, core.inds, outpath="corefit_structures")
rf <- rmsf(xyz)
plotb3(rf, sse=pdb)
abline(v=100, col="gray", ylab="RMSF")
Predicted Alignment Error
library(jsonlite)
# list all PAE JSON files (should be 5)
pae_files <- list.files(pattern=".*model.*\\.json$", full.names=TRUE)
basename(pae_files)
[1] "hivpr_23119_scores_rank_001_alphafold2_multimer_v3_model_4_seed_000.json"
[2] "hivpr_23119_scores_rank_002_alphafold2_multimer_v3_model_1_seed_000.json"
[3] "hivpr_23119_scores_rank_003_alphafold2_multimer_v3_model_5_seed_000.json"
[4] "hivpr_23119_scores_rank_004_alphafold2_multimer_v3_model_2_seed_000.json"
[5] "hivpr_23119_scores_rank_005_alphafold2_multimer_v3_model_3_seed_000.json"
length(pae_files)
[1] 5
pae1 <- read_json(pae_files[1], simplifyVector = TRUE)
pae5 <- read_json(pae_files[5], simplifyVector = TRUE)
attributes(pae1)
$names
[1] "plddt" "max_pae" "pae" "ptm" "iptm"
head(pae1$plddt)
[1] 90.81 93.25 93.69 92.88 95.25 89.44
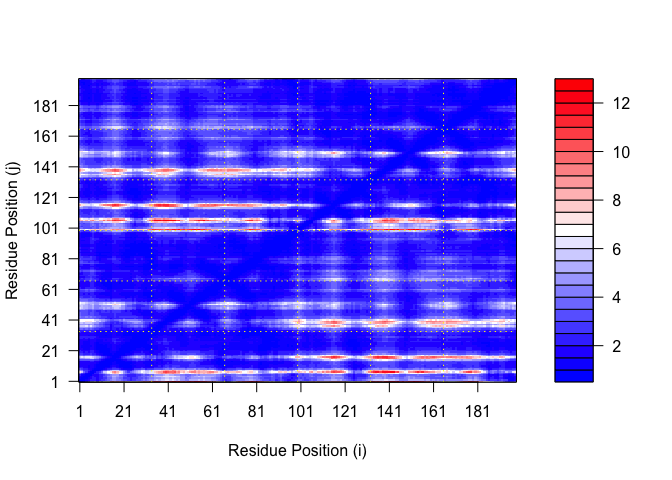
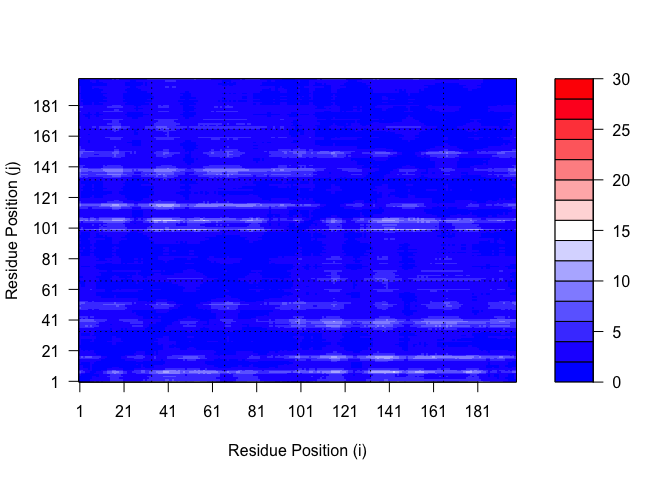
pae1$max_pae
[1] 12.84375
pae5$max_pae
[1] 29.59375
library(bio3d)
plot.dmat(pae1$pae,
xlab="Residue Position (i)",
ylab="Residue Position (j)")
plot.dmat(pae5$pae,
xlab="Residue Position (i)",
ylab="Residue Position (j)",
grid.col = "black",
zlim=c(0,30))
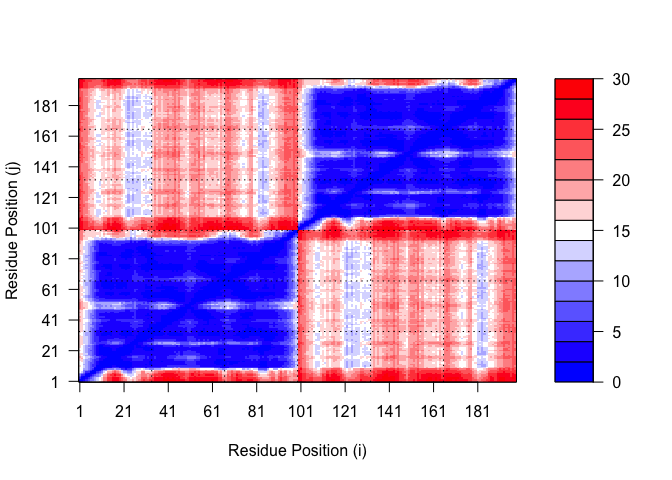
plot.dmat(pae1$pae,
xlab="Residue Position (i)",
ylab="Residue Position (j)",
grid.col = "black",
zlim=c(0,30))
Residue conservation from alignment file
aln_file <- list.files(pattern="\\.a3m$", full.names=TRUE)
aln_file
[1] "./hivpr_23119.a3m"
library(bio3d)
aln <- read.fasta(aln_file[1], to.upper = TRUE)
[1] " ** Duplicated sequence id's: 101 **"
[2] " ** Duplicated sequence id's: 101 **"
dim(aln$ali)
[1] 5397 132
pdb <- read.pdb(pdb_files[1])
sim <- conserv(aln)
plotb3(sim[1:99],
sse = trim.pdb(pdb, chain = "A"),
ylab = "Conservation Score")
Warning in pdb2sse(sse): No helix and sheet defined in input 'sse' PDB object:
try using dssp()
Warning in plotb3(sim[1:99], sse = trim.pdb(pdb, chain = "A"), ylab =
"Conservation Score"): Length of input 'sse' does not equal the length of input
'x'; Ignoring 'sse'
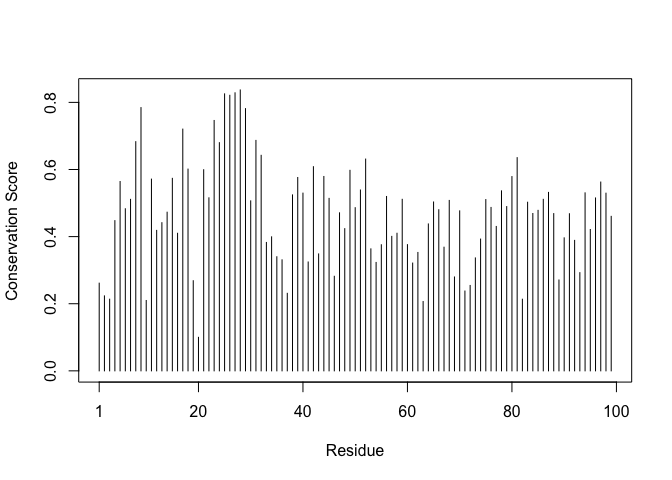
con <- consensus(aln, cutoff = 0.9)
con$seq
[1] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[19] "-" "-" "-" "-" "-" "-" "D" "T" "G" "A" "-" "-" "-" "-" "-" "-" "-" "-"
[37] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[55] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[73] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[91] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[109] "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-" "-"
[127] "-" "-" "-" "-" "-" "-"
m1.pdb <- read.pdb(pdb_files[1])
occ <- vec2resno(c(sim[1:99], sim[1:99]), m1.pdb$atom$resno)
write.pdb(m1.pdb, o = occ, file = "m1_conserv.pdb")